James A. Fellows Yates 0000-0001-5585-6277
· jfy133
· jafellowsyates
Microbiome Sciences Group, Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany; Institut für Vor- und Frühgeschichtliche Archäologie und Provinzialrömische Archäologie, Ludwig Maximilian University, Munich, Germany
· Funded by Max Planck Society
Thiseas C. Lamnidis 0000-0003-4485-8570
· TCLamnidis
· TCLamnidis
Population Genetics Group, Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany
· Funded by Max Planck Society
Maxime Borry 0000-0001-9140-7559
· maxibor
· notmaxib
Microbiome Sciences Group, Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany
· Funded by Max Planck Society
Aida Andrades Valtueña 0000-0002-1737-2228
· aidaanva
· aidaanva
Computational Pathogenomics Group, Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany
· Funded by Max Planck Society
Zandra Fagernäs 0000-0003-2667-3556
· ZandraFagernas
· ZandraSelina
Microbiome Sciences Group, Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany
· Funded by Max Planck Society
Stephen Clayton 0000-0001-5223-9695
· sc13-bioinf
Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany
· Funded by Max Planck Society
Maxime U. Garcia 0000-0003-2827-9261
· MaxUlysse
· gau
Department of Oncology-Pathology, Karolinska Institutet, Stockholm, Sweden
· Funded by Barncancerfonden
Alexander Peltzer 0000-0002-6503-2180
· apeltzer
· alex_peltzer
Quantitative Biology Center (QBiC), Eberhard-Karls-Universität, Tübingen, Germany; Department of Archaeogenetics, Max Planck Institute for the Science of Human History, Jena, Germany
Abstract
The broadening utilisation of ancient DNA to address archaeological,
palaeontological, and biological questions is resulting in a rising diversity in
the size of laboratories and scale of analyses being performed. In the context
of this heterogeneous landscape, we present nf-core/eager, an advanced and
entirely redesigned and extended version of the EAGER pipeline for the analysis of ancient genomic
data. This Nextflow pipeline aims to address three main themes: accessibility
and adaptability to different computing configurations, reproducibility to
ensure robust analytical standards, and updating the pipeline to
the latest routine ancient genomic practises. This new version of EAGER has been
developed within the nf-core initiative to ensure high-quality software
development and maintenance support; contributing to a long-term lifecycle for
the pipeline. nf-core/eager will assist in ensuring that ancient DNA sequencing
data can be used by a diverse range of research groups and fields.
Introduction
Ancient DNA (aDNA) has become a widely accepted source of biological data,
helping to provide new perspectives for a range of fields including archaeology,
cultural heritage, evolutionary biology, ecology, and palaeontology. The
utilisation of short-read high-throughput sequencing has allowed the recovery of
whole genomes and genome-wide data from a wide variety of sources, including
(but not limited to), the skeletal remains of animals
[1,2,3,4], modern and archaic
humans [5,6,7,8], bacteria
[9,10,11], viruses [12,13], plants [14,15], palaeofaeces [16,17], dental calculus [18,19], sediments [20,21], medical slides [22],
parchment [23], and recently, ancient ‘chewing gum’
[24,25]. Improvement
in laboratory protocols to increase yields of otherwise trace amounts of DNA has
at the same time led to studies that can total hundreds of ancient individuals
[26,27], spanning single
[28] to thousands of organisms [18].
These differences of disciplines have led to a heterogeneous landscape in terms
of the types of analyses undertaken, and their computational resource
requirements [29,30]. Taking
into consideration the unequal distribution of resources (and infrastructure
such as internet connection), easy-to-deploy, streamlined and efficient
pipelines can help increase accessibility to high-quality analyses.
The degraded nature of aDNA poses an extra layer of complexity to standard
modern genomic analysis. Through a variety of processes [31]
DNA molecules fragment over time, resulting in ultra-short molecules
[32]. These sequences have low nucleotide complexity
making it difficult to identify with precision which part of the genome a read
(a sequenced DNA molecule) is derived from. Fragmentation without a ‘clean
break’ leads to uneven ends, consisting of single-stranded ‘overhangs’ at end of
molecules, which are susceptible to chemical processes such as deamination of
nucleotides. These damaged nucleotides then lead to misincorporation of
complementary bases during library construction for high-throughput DNA
sequencing [33]. On top of this, taphonomic processes
such as heat, moisture, and microbial- and burial-environment processes lead to
varying rates of degradation [34,35]. The original DNA content of a sample
is therefore increasingly lost over time and supplanted by younger
‘environmental’ DNA. Later handling by archaeologists, museum curators, and
other researchers can also contribute ‘modern’ contamination. While these
characteristics can help provide evidence towards the ‘authenticity’ of true
aDNA sequences (e.g. the aDNA cytosine to thymine or C to T ‘damage’
deamination profiles [36]), they also pose
specific challenges for genome reconstruction, such as unspecific DNA alignment
and/or low coverage and miscoding lesions that can result in low-confidence
genotyping. These factors often lead to prohibitive sequencing costs when
retrieving enough data for modern high-throughput short-read sequencing data
pipelines (such as more than 1 billion reads for a 1X depth coverage Yersinia
pestis genome [37]), and thus aDNA-tailored
methods and techniques are required to overcome these challenges.
Two previously published and commonly used pipelines in the field are PALEOMIX
[38] and EAGER [39]. These
two pipelines take a similar approach to link together standard tools used for
Illumina high-throughput short-read data processing (sequencing quality control,
sequencing adapter removal/and or paired-end read merging, mapping of reads to a
reference genome, genotyping, etc.). However, they have a specific focus on
tools that are designed for, or well-suited for aDNA (such as the bwa aln
algorithm for ultra-short molecules [40] and
mapDamage [41] for evaluation of aDNA
characteristics). Yet, neither of these genome reconstruction pipelines have had
major updates to bring them in-line with current routine aDNA analyses.
Metagenomic screening of off-target genomic reads for pathogens or microbiomes
[18,19] has become particularly common
in palaeo- and archaeogenetics, given its role in revealing widespread
infectious disease and possible epidemics that have sometimes been previously
undetected in the archaeological record [12,13,37,42]. Without easy access to the latest field-established
analytical routines, ancient genomic studies risk being published without the
necessary quality control checks that ensure aDNA authenticity, as well as
limiting the full range of possibilities from their data. Given that material
from samples is limited, there are both ethical as well as economical interests
to maximise analytical yield [43].
To address these shortcomings, we have completely re-implemented the latest
version of the EAGER pipeline in Nextflow [44] (a
domain-specific-language or ‘DSL’, specifically designed for the construction of
omics analysis pipelines), introduced new features, and more flexible pipeline
configuration. In addition, the renamed pipeline - nf-core/eager
- has been developed in the context of the nf-core community framework
[45], which enforces strict guidelines for
best-practices in software development.
Results and Discussion
Scalability, Portability, and Efficiency
The re-implementation of EAGER into Nextflow offers a range of benefits over the
original custom pipeline framework.
Firstly, the new framework provides immediate integration of nf-core/eager into
various job schedulers in POSIX High-Performance-Cluster (HPC) environments,
cloud computing resources, as well as local workstations. This portability
allows users to set up nf-core/eager regardless of the type of computing
infrastructure or cluster size (if applicable), with minimal effort or
configuration. This facilitates reproducibility and therefore maintenance of
standards within the field. Portability is further assisted by the in-built
compatibility with software environments and containers such as Conda
[46], Docker [47] and Singularity
[48]. These are isolated software ‘sandbox’ environments
that include all software (with exact versions) required by the pipeline, in a
form that is installable and runnable by users regardless of the set up of their
local software environment. Another major change with nf-core/eager is that the
primary user interaction mode of a pipeline run set up is now with a
command-line interface (CLI), replacing the graphical-user-interface (GUI) of
the original EAGER pipeline. This is more portable and compatible with most HPCs (that
may not offer display of a window system), and is in line with the vast majority
of bioinformatics tools. We therefore believe this will not be a hindrance to
new researchers from outside computational biology. However, a GUI-based
pipeline set up is still avaliable via the nf-core website’s Launch page
[49], which provides a common
GUI format across multiple pipelines, as well as additional robustness checks of
input parameters for those less familiar with CLIs. Typically the output of the
launch functionality is a JSON file that can be used with a nf-core/tools launch
command as a single parameter (similar to the original EAGER), however
integration with Nextflow’s companion monitoring tool tower.nf
[50] also allows direct submission of pipelines without any
command line usage.
Secondly, reproducibility is made easier through the use of ‘profiles’ that can
define configuration parameters. These profiles can be managed at different
hierarchical levels. HPC-level profiles can specify parameters for the computing
environment (job schedulers, cache locations for containers, maximum memory and
CPU resources etc.), which can be centrally managed to ensure all users of a
group use the same settings. Pipeline-level profiles, specifying parameters for
nf-core/eager itself, allow fast access to routinely-run pipeline parameters via a
single flag in the nf-core/eager run command, without having to configure each
new run from scratch. Compared to the original EAGER, which utilised per-FASTQ
XML files with hardcoded filepaths for a specific user’s server, nf-core/eager
allows researchers to publish the specific profile used in their runs alongside
their publications, that can also be used by other groups to generate the same
results. Usage of profiles can also reduce mistakes caused by insufficient
‘prose’ based reporting of program settings that can be regularly found in the
literature. The default nf-core/eager profile uses parameters evaluated in
different aDNA-specific contexts (e.g. in [51]), and
will be updated in each new release as new studies are published.
Finally, nf-core/eager provides improved efficiency over the original EAGER
pipeline by replacing sample-by-sample sequential processing with Nextflow’s
asynchronous job parallelisation, whereby multiple pipeline steps and samples
are run in parallel (in addition to natively parallelised pipeline steps). This
is similar to the approach taken by PALEOMIX, however nf-core/eager expands this
by utilising Nextflow’s ability to customise the resource parameters for every
job in the pipeline; reducing unnecessary resource allocation that can occur
with unfamiliar users to each step of a high-throughput short-read data
processing pipeline. This is particularly pertinent given the increasing use of
centralised HPCs or cloud computing that often use per-hour cost calculations.
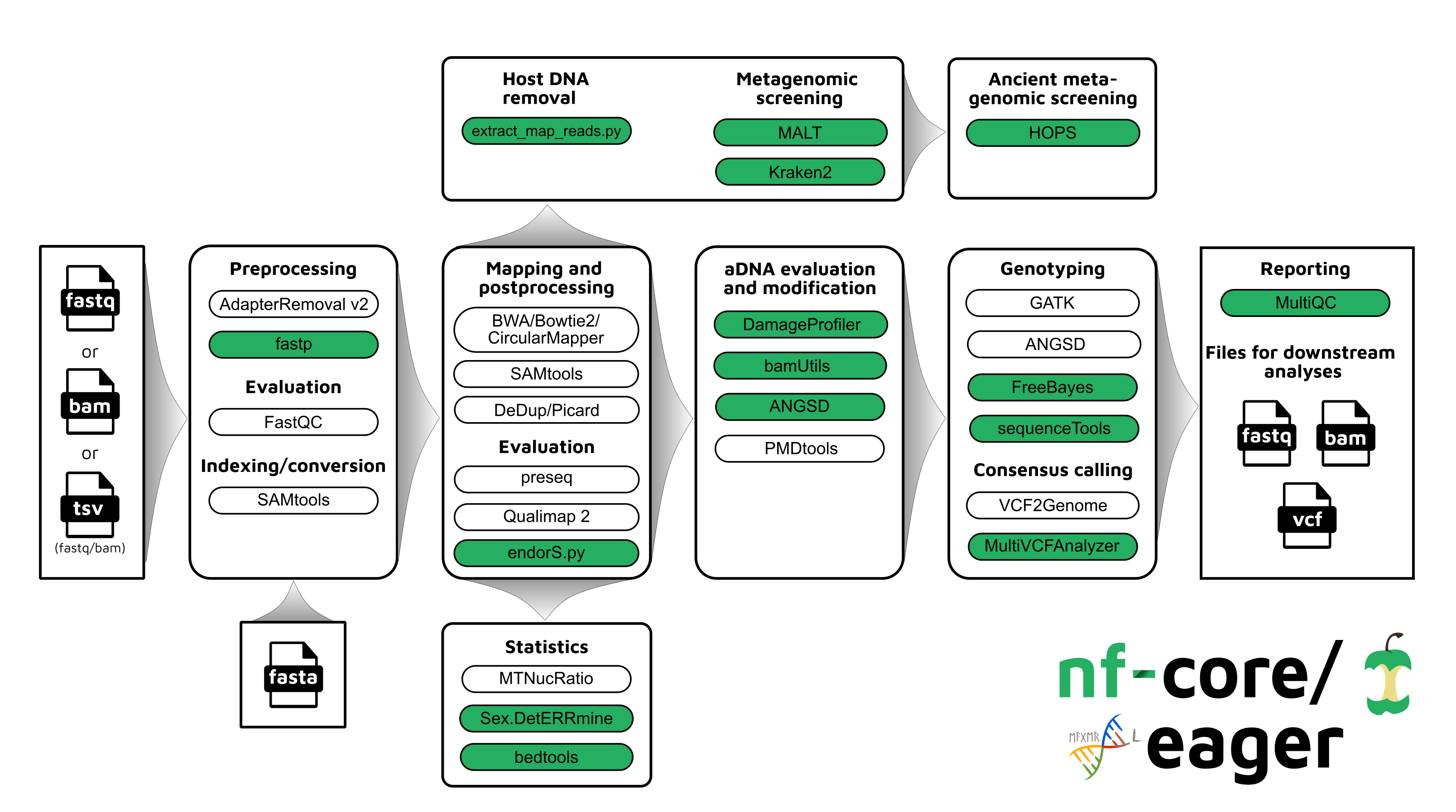
Updated Workflow
nf-core/eager follows a similar structural foundation to the original version of
EAGER and partially to PALEOMIX. Given Illumina short-read FASTQ and/or BAM
files and a reference FASTA file, the core functionality of nf-core/eager can be split in five
main stages:
In nf-core/eager, all tools originally used in EAGER have been updated to their
latest versions, as available on Bioconda [70] and
Conda-forge [71], to ensure widespread
accessibility and stability of utilised tools. The mapDamage2 (for damage
profile generation) [36] and Schmutzi (for
mitochondrial contamination estimation) [72] methods
have not been carried over to nf-core/eager, the first because a more performant
successor method is now available (DamageProfiler), and the latter because a
stable release of the method could not be migrated to Bioconda. We anticipate
that there will be an updated version of Schmutzi in the near future that will
allow us to integrate the method again into nf-core/eager. As an alternative,
estimation of human nuclear contamination is now offered through
ANGSD [67]. Support for the Bowtie2 aligner
[57] has been updated to have default settings optimised
for aDNA [73].
New tools to the basic workflow include fastp
[54] for the removal of ‘poly-G’ sequencing
artefacts that are common in 2-colour Illumina sequencing machines (such as the
increasingly popular NextSeq and NovaSeq platforms
[74]).
For variant calling, we have now included FreeBayes [75] as an
alternative to the human-focused GATK tools, and have also added pileupCaller
[68] for generation of genotyping
formats commonly utilised in ancient human population analysis. We have also
maintained the possibility of using the now unsupported GATK UnifiedGenotyper,
as the supported replacement, GATK HaplotypeCaller, performs de novo assembly
around possible variants; something that may not be suitable for low-coverage
aDNA data.
Figure 1: Simplified schematic of the nf-core/eager workflow pipeline. Green filled
bubbles indicate new functionality added over the original EAGER
pipeline.
Additional functionality tailored for ancient bacterial genomics includes
integration of a SNP alignment generation tool, MultiVCFAnalyzer
[9], which includes the ability to make an assessment of
levels of cross-mapping from different related taxa to a reference genome - a
common challenge in ancient bacterial genome reconstruction
[35]. The output SNP consensus alignment
FASTA file can then be used for downstream analyses such as phylogenetic tree
construction. Simple coverage statistics of particular annotations (e.g. genes)
of an input reference is offered by bedtools
[62], which can be used in cases such as for
providing initial indications of functional differences between ancient
bacterial strains (as in [42]). When using a human
reference genome, nf-core/eager can also give estimates of the relative coverage
on the X and Y chromosomes with Sex.DetERRmine that can be used to infer the
biological sex of a given human individual [63]. A
dedicated ‘endogenous DNA’ calculator (endorS.py) is also included, to provide a
percentage estimate of the sequenced reads matching the reference (‘on-target’)
from the total number of reads sequenced per library.
Given the large amount of sequencing often required to yield sufficient genome
coverage from aDNA data, palaeogeneticists tend to use multiple (differently
treated) libraries, and/or merge data from multiple sequencing runs of each
library or even samples. The original EAGER pipeline could only run a single
library at a time, and in these contexts required significant manual user input
in merging different FASTQ or BAM files of related libraries. A major upgrade in
nf-core/eager is that the new pipeline supports automated processing of complex
sequencing strategies for many samples, similar to PALEOMIX. This is facilitated
by the optional use of a simple table (in TSV format, a format more
commonly used in wet-lab stages of data generation, compared to PALEOMIX’s YAML
format) that includes file paths and additional metadata such as sample name,
library name, sequencing lane, colour chemistry, and UDG treatment. This allows
automated and simultaneous processing and appropriate merging and treatment of
heterogeneous data from multiple sequencing runs and/or library types.
The original EAGER and PALEOMIX pipelines required users to look through many
independent output directories and files to make full assessment of their
sequencing data. This has now been replaced in nf-core/eager with a much more
extensive MultiQC report [69]. This tool
aggregates the log files of every supported tool into a single interactive
report, and assists users in making a fuller assessment of their sequencing and
analysis runs. We have developed a corresponding MultiQC module for every tool
used by nf-core/eager, where possible, to enable comprehensive evaluation of all
stages of the pipeline.
We have further extended the functionality of the original EAGER pipeline by
adding ancient metagenomic analysis; allowing reconstruction of the wider
taxonomic content of a sample. We have added the possibility to screen all
off-target reads (not mapped to the reference genome) with two metagenomic
profilers: MALT [76,77] and
Kraken2 [78], in parallel to the mapping to a given
reference genome (typically of the host individual, assuming the sample is a
host organism). Characterisation of properties of authentic aDNA from
metagenomic MALT alignments is carried out with MaltExtract of the HOPS pipeline
[79]. This functionality can be used either for
microbiome screening or putative pathogen detection. Ancient metagenomic studies
sometimes include comparative samples from living individuals
[80]. To support open data, whilst respecting
personal data privacy, nf-core/eager includes a ‘FASTQ host removal’ script that
creates raw FASTQ files, but with all reads successfully mapped to the reference
genome removed. This allows for safe upload of metagenomic non-host sequencing
data to public repositories after removal of identifiable (human) data, for
example for microbiome studies.
An overview of the entire pipeline is shown in Figure 1,
and a tabular comparison of functionality between EAGER, PALEOMIX and
nf-core/eager is in Table 1.
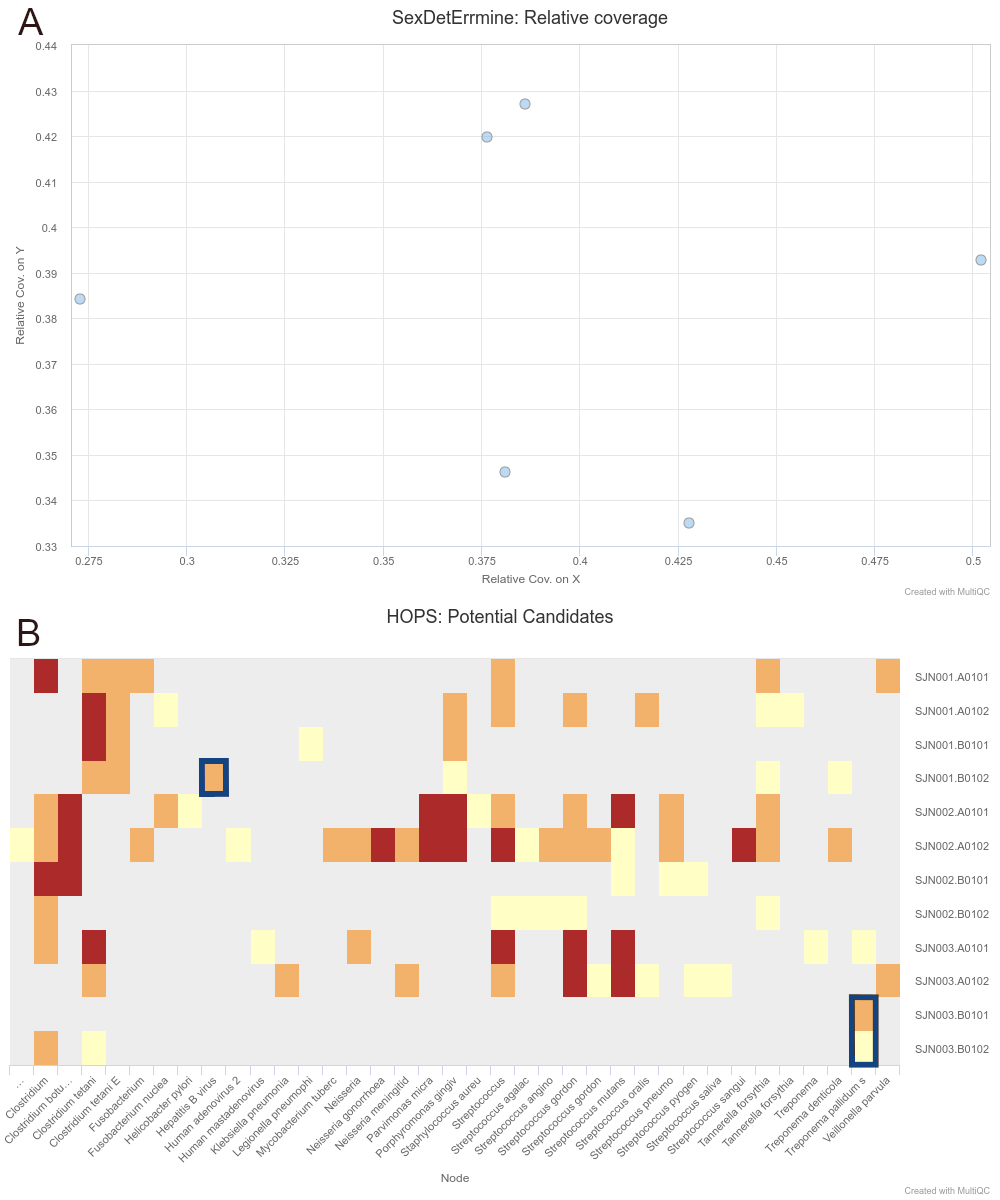
To demonstrate the simultaneous genomic analysis of human DNA and metagenomic
screening for putative pathogens, as well as improved results reporting, we
re-analysed data from Barquera et al. 2020 [81], who
performed a multi-discipline study of three 16th century individuals excavated
from a mass burial site in Mexico City. The authors reported genetic results
showing sufficient on-target human DNA (>1%) with typical aDNA damage (>20% C to
T reference mismatches in the first base of the 5’ ends of reads) for downstream
population-genetic analysis and Y-chromosome coverage indicative that the three
individuals were genetically male. In addition, one individual (Lab ID: SJN003)
contained DNA suggesting a possible infection by Treponema pallidum, a species
with a variety of strains that can cause diseases such as syphilis, bejel and
yaws, and a second individual (Lab ID: SJN001) displayed reads similar to the
Hepatitis B virus. Both results were confirmed by the authors via in-solution
enrichment approaches.
We were able to successfully replicate the human and pathogen screening results
in a single run of nf-core/eager. Mapping to the human reference genome (hs37d5)
with BWA aln and binning of off-target reads with MALT to the NCBI Nucleotide
database (2017-10-26), yielded the same results of all individuals having a
biological sex of male, as well as the same frequency of C to T miscoding
lesions and short fragment lengths (both characteristic of true aDNA).
Metagenomic hits to both pathogens from the corresponding individuals that also
yielded complete genomes in the original publication were also detected. Both
results and other processing statistics were identified via a single interactive
MultiQC report, excerpts of which can be seen in Figure
2. The full interactive report can be seen in the
supplementary information.
Figure 2: Sections of a MultiQC report (v1.10dev) with the outcome of simultaneous human
DNA and microbial pathogen screening with nf-core/eager, including A
Sex.DetERRmine output of biological sex assignment with coverages on X and Y
being half of that of autosomes, indicative of male individuals, and B HOPS
output with positive detection of both Treponema pallidum and Hepatitis B
virus reads - indicated with blue boxes. Other taxa in HOPS output represent
typical environmental contamination and oral commensal microbiota found in
archaeological teeth. Data was Illumina shotgun sequencing data from Barquera et
al. 2020 [81], and replicated results here were
originally verified in the publication via enrichment methods. The full
interactive reports for both MultiQC v1.9 and v1.10 (see methods) can be seen in
the supplementary
information.
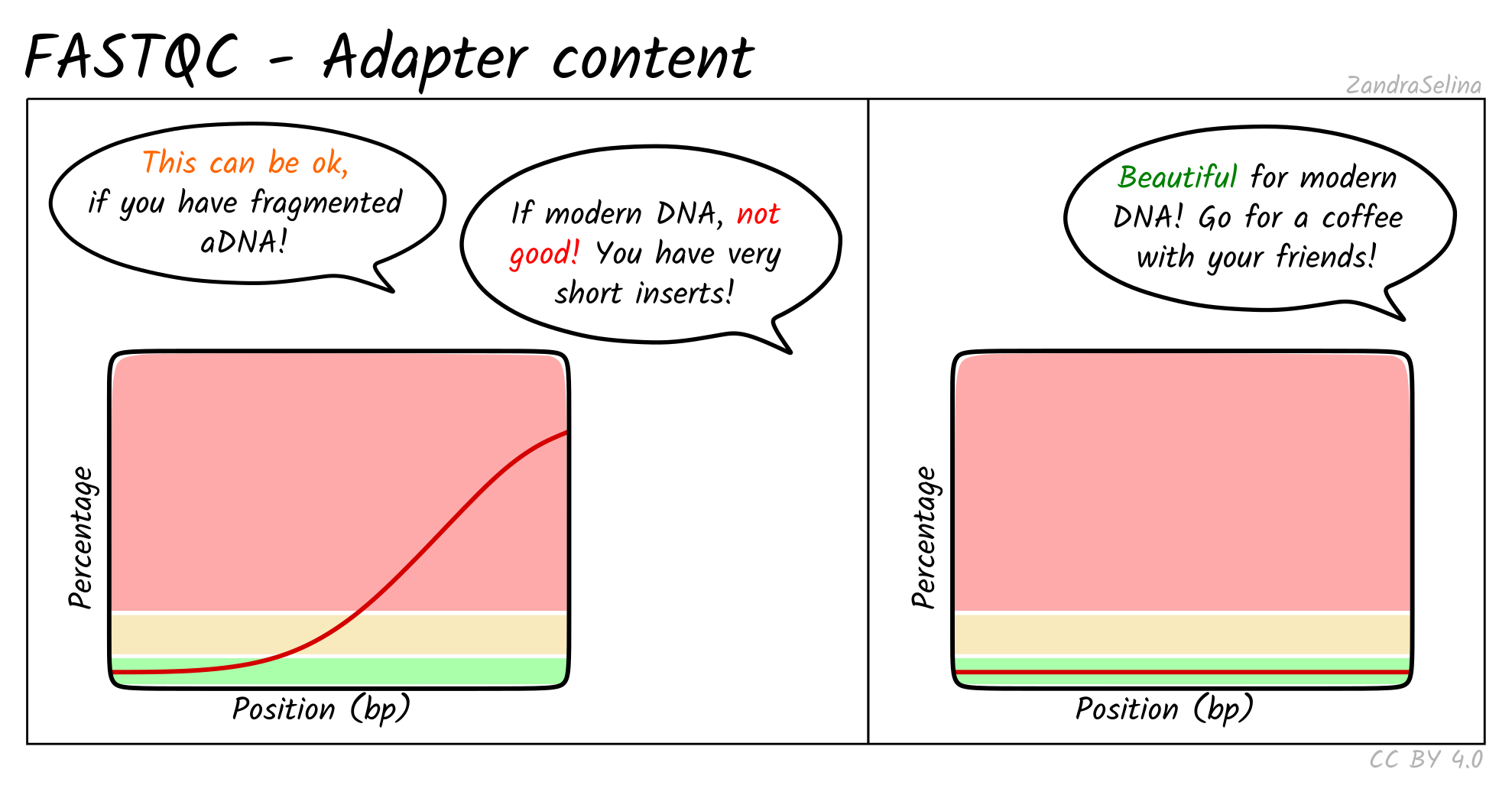
Accessibility
Alongside the interactive MultiQC report, we have written extensive
documentation on all parts of running and interpreting the output of the
pipeline. Given that a large fraction of aDNA researchers come from fields
outside computational biology, and thus may have limited computational training,
we have written documentation and tutorials [82]
that also gives guidance on how to run the pipeline and interpret each section
of the report in the context of high-throughput sequencing data, but with with a
special focus on aDNA. This includes best practice or expected output schematic
images that are published under CC-BY licenses to allow for use in other
training material (an example can be seen in Figure 3).
We hope this open-access resource will make the study of aDNA more accessible to
researchers new to the field, by providing practical guidelines on how to
evaluate characteristics and effects of aDNA on downstream analyses.
Figure 3: Example schematic images of pipeline output documentation that can assist new
users in the interpretation of high-throughput sequencing aDNA
processing.
The development of nf-core/eager in Nextflow and the nf-core initiative will
also improve open-source development, while ensuring the high quality of
community contributions to the pipeline. While Nextflow is written primarily in
Groovy, the Nextflow DSL simplifies a number of concepts to an intermediate
level that bioinformaticians without Java/Groovy experience can easily access
(regardless of own programming language experience). Furthermore, Nextflow
places ubiquitous and more widely known command-line interfaces, such as bash,
in a prominent position within the code, rather than custom Java code and
classes (as in EAGER). We hope this will motivate further bug fixes and
feature contributions from the community, to keep the pipeline state-of-the-art
and ensure a longer life-cycle. This will also be supported by the open and
active nf-core community who provide general guidance and advice on developing
Nextflow and nf-core pipelines.
Comparisons with other pipelines
The scope of nf-core/eager is as a generic, initial data processing and
screening tool, and not to act as a tool for performing more experimental
analyses that requires extensive parameter testing such as modelling. As such,
while similar pipelines designed for aDNA have also been released, for example
ATLAS [83], these generally have been designed with specific
contexts in mind (e.g. human population genetics). We therefore have opted to
not include common downstream analysis such as Principal Component Analysis for
population genetics, or phylogenetic analysis for microbial genomics, but rather
focus on ensuring nf-core/eager produces useful files that can be easily used as
input for common but more experimental and specialised downstream analysis.
Therefore, we compared pipeline run-times of two functionally equivalent and
previously published pipelines to show that the new implementation of
nf-core/eager is equivalent or more efficient than EAGER or PALEOMIX.
Table 1: Comparison of pipeline functionality of common ancient DNA processing
pipelines. Tick represents full functionality, tilde represents partial functionality, and cross represents not implemented.
Category
Functionality
EAGER
PALEOMIX
nf-core/eager
Infrastructure
Reproducible software environments offered
✓
✗
✓
HPC scheduler integration
✗
✗
✓
Cloud computing integration
✗
✗
✓
Per-process resource optimisation
✗
~
✓
Pipeline-step parallelisation
✗
✓
✓
Command line set up
✗
✓
✓
GUI set up
✓
✗
✓
Preprocessing
Sequencing lane merging
✓
✓
✓
Sequencing quality control
✓
✗
✓
Sequencing artefact removal
✗
✗
✓
Adapter clipping/read merging
✓
✓
✓
Post-processing sequencing QC
✗
✗
✓
Alignment
Reference mapping
✓
✓
✓
Reference mapping statistics
✓
✓
✓
Multi-reference mapping
✗
✓
✗
Postprocessing
Mapped reads filtering
✓
✓
✓
Off-target metagenomic profiling
✗
✗
✓
Off-target metagenomic authentication
✗
✗
✓
Library complexity estimation
✓
✗
✓
Duplicate removal
✓
✗
✓
BAM merging
✗
✓
✓
Authentication
Damage read filtering
✓
✗
✓
Contamination estimation (Human)
✓
✗
✓
Biological sex determination (Human)
✗
✗
✓
Genome coverage estimation
✓
✓
✓
Damage calculation
✓
✓
✓
Damage rescaling
✗
✓
~
Downstream
SNP Calling/Genotyping
✓
~
✓
Consensus sequence generation
✓
~
✓
Regions of interest statistics
~
✓
✓
We ran each pipeline on a subset of Viking-age genomic data of cod (Gadus
morhua) from Star et al. 2017 [4]. This data was
originally run using PALEOMIX, and was re-run here as described, but with the
latest version of PALEOMIX (v1.2.14), and with equivalent settings for the other
two pipelines as close as possible to the original paper (EAGER with v1.92.33,
and nf-core/EAGER with v2.2.0dev, commit
830c22d). The respective benchmarking
environment and exact pipeline run settings can be seen in the Methods and
Supplementary Information. Two samples each with three Illumina paired-end
sequencing runs were analysed, with adapter clipping and merging
(AdapterRemoval), mapping (BWA aln), duplicate removal (Picard’s MarkDuplicates)
and damage profiling (PALEOMIX: mapDamage2, EAGER and nf-core/EAGER:
DamageProfiler) steps being performed. We ran the commands for each tool
sequentially, but repeated these batches of commands 10 times - to account for
variability in the cloud service’s IO connection. Run times were measured using
the GNU time tool (v1.7).
Table 2: Comparison of run times in minutes between three ancient DNA pipelines.
PALEOMIX and nf-core/eager have additional runs with ‘optimised’ parameters with
fairer computational resources matching modern multi-threading strategies.
Values represent mean and standard deviation of run times in minutes, calculated
from the output of the GNU time tool. Real: real time, System: cumulative CPU system-task times,
User: cumulative CPU time of all tasks.
Pipeline
Version
Environment
real
sys
user
nf-core-eager (optimised)
2.2.0dev
singularity
105.6 ± 4.6
13.6 ± 0.7
1593 ± 79.7
PALEOMIX (optimised)
1.2.14
conda
130.6 ± 8.7
12 ± 0.7
1820.2 ± 36.9
nf-core-eager
2.2.0dev
singularity
209.2 ± 4.4
11 ± 0.9
1407.7 ± 30.2
EAGER
1.92.37
singularity
224.2 ± 4.9
22.9 ± 0.3
1736.3 ± 70.2
PALEOMIX
1.2.14
conda
314.6 ± 2.9
10.7 ± 1
1506.7 ± 14
A summary of runtimes of the benchmarking tests can be seen in Table
2. nf-core/eager showed fastest runtimes across all
three time metrics when running on default parameters. This
highlights the improved efficiency of nf-core/eager’s asynchronous processing
system and per-process resource customisation (here represented by nf-core/eager
defaults designed for typical HPC set ups).
As a more realistic demonstration of modern computing multi-threading set ups,
we also re-ran PALEOMIX with the flag –max-bwa-threads set to 4 (listed in
Table 2 as ‘optimised’), which is equivalent to a
single BWA aln process of nf-core/eager. This resulted in a much faster run-time
than that of default nf-core/eager, due to the approach of PALEOMIX of mapping
each lane of a library separately, whereas nf-core/eager will map all lanes of a
single library merged together. Therefore, given that each library was split
across three lanes, increasing the threads of BWA aln to 4 resulted in 12 per
library, whereas nf-core/eager only gave 4 (by default) for a single BWA aln
process of one library. While the PALEOMIX approach is valid, we opted to retain
the per-library mapping as it is often the longest running step of
high-throughput sequencing genome-mapping pipelines, and it prevents flooding of
HPC scheduling systems with many long-running jobs. Secondly, if users regularly
use multi-lane data, due to nf-core/eager’s fine-granularity control, they can
simply modify nf-core/eager’s BWA aln process resources via config files to
account for this. When we optimised parameters that were used for BWA aln’s
multi-threading, and the number of multiple lanes to the same number of BWA aln
threads as the optimised PALEOMIX run, nf-core/eager again displayed faster
runtimes. All metrics including mapped reads, percentage on-target, mean depth
coverage and mean read lengths across all pipelines were extremely similar
across all pipelines and replicates (see methods and Table
3).
Conclusion
nf-core/eager is an efficient, portable, and accessible pipeline for processing
and screening ancient (meta)genomic data. This re-implementation of EAGER into
Nextflow and nf-core will improve reproducibility and scalability of rapidly
increasing aDNA datasets, for both large and small laboratories. Extensive
documentation also enables newcomers to the field to get a practical
understanding on how to interpret aDNA in the context of NGS data processing.
Ultimately, nf-core/eager provides easier access to the latest tools and routine
screening analyses commonly used in the field, and sets up the pipeline for
remaining at the forefront of palaeogenetic analysis.
Methods
Installation
nf-core/eager requires only three dependencies: Java (version >= 8), Nextflow,
and either a functional Conda installation or Docker/Singularity engine
installation. A quick installation guide to follow to get started can be found
in the Quick start section of the nf-core/eager repository
[84].
Running
After installation, users can run the pipeline using standard test data by
utilising some of the test profiles we provide (e.g. using Docker):
nextflow run nf-core/eager -r 2.2.0 -profile test_tsv,docker
This will download test data automatically (as recorded in the test_tsv
profile), run the pipeline locally with all software tools containerised in a
Docker image. The pipeline will store the output of that run in the default
‘./results’ folder of the current directory.
The default pipeline settings assumes paired-end FASTQ data, and will run:
DamageProfiler and Qualimap2 (for genome coverage statistics)
MultiQC pipeline run report
If no additional FASTA indices are given, these will also be generated.
The pipeline is highly configurable and most modules can be turned on-and-off
using different flags at the request of the user, to allow a high level of
customisation to each user’s needs. For example, to include metagenomic
screening of off-target reads, and sex determination based on on-target mappings
of pre-clipped single-end data:
In addition to private locally defined profiles, we utilise a central
configuration repository to enable users from various institutions to use
pipelines on their particular infrastructure more easily
[85]. There are multiple resources listed
in this repository with information on how to add a user’s own institutional
configuration profile with help from the nf-core community. These profiles can
be both generic for all nf-core pipelines, but also customised for specific
pipelines.
Users can customise this infrastructure profile by themselves, with the nf-core
community, or with their local system administrator to make sure that the
pipeline runs successfully, and can then rely on the Nextflow and nf-core
framework to ensure compatibility upon further infrastructure changes. For
example, in order to run the nf-core/eager pipeline at the Max Planck Institute
for the Science of Human History (MPI-SHH), users only have to run:
nextflow run nf-core/eager -r 2.2.0 -profile test_tsv,sdag,shh
This runs the testing profile of the nf-core/eager pipeline with parameters
specifically adapted to a specific HPC system at the MPI-SHH. In some cases,
similar institutional configs for other institutions may already exist
(originally utilised for different nf-core pipelines), so users need not
necessarily write their own.
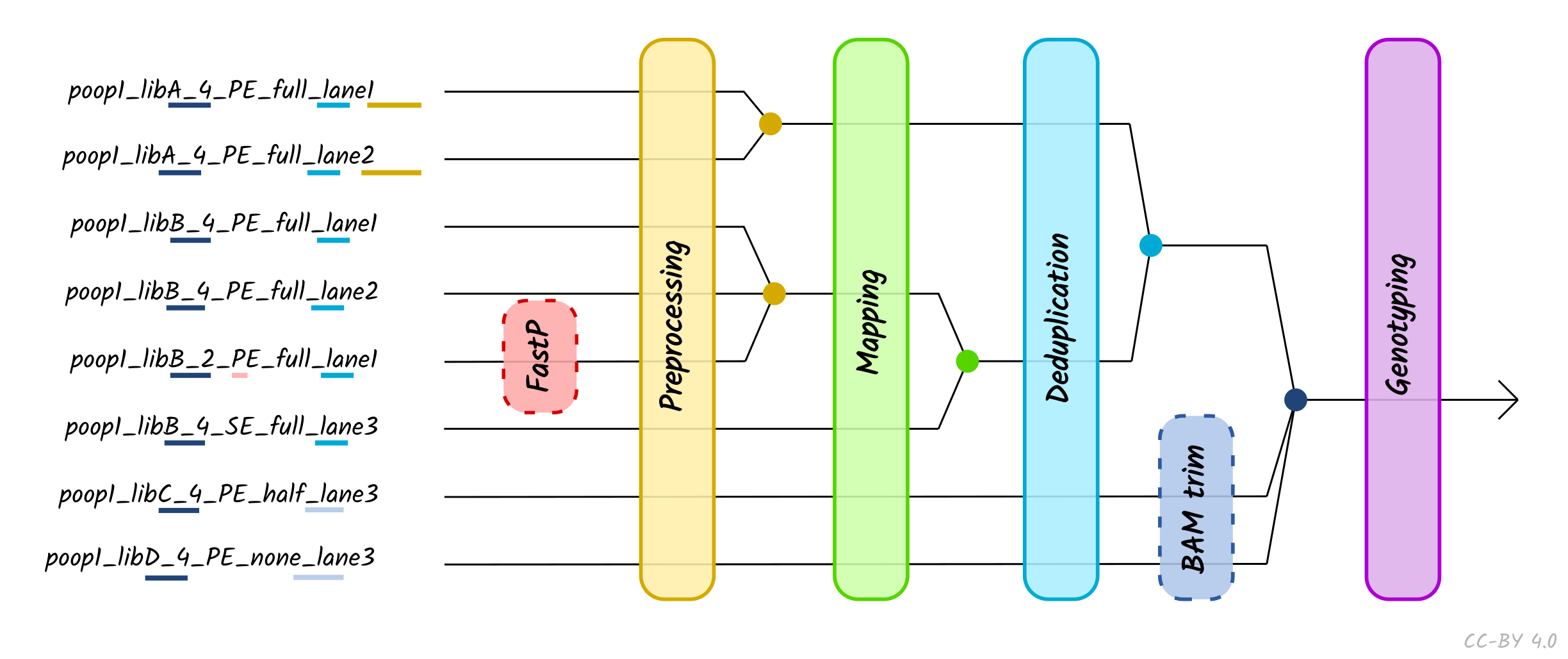
Inputs
The pipeline can be started using (raw) FASTQ files from sequencing or
pre-mapped BAM files. Additionally, the pipeline requires a FASTA reference
genome. If BAM input is provided, an optional conversion to FASTQ is offered,
otherwise BAM files processing will start from the post-mapping stage.
If users have complex set-ups, e.g. multiple sequencing lanes that require
merging of files, the pipeline can be supplied with a tab separated value (TSV)
file to enable such complex data handling. Both FASTQs and BAMs can be provided
in this set up. FASTQs with the same library name and sequencing chemistry but
sequenced across multiple lanes will be concatenated after adapter removal and
prior mapping. Libraries with different sequencing chemistry kits (paired- vs.
single-end) will be merged after mapping. Libraries with the same sample name
and with the same UDG treatment, will be merged after deduplication. If
libraries with the sample name have different UDG treatment, these will be
merged after the aDNA modification stage (i.e. BAM trimming or PMDtools, if
turned on), prior to genotyping, as shown in Figure 4.
Figure 4: Schematic of different processing and merging points based on the nature of
different libraries, as specified by the metadata of a TSV file. Dashed boxes
represent optional library-specific
processes. Colours refer to each merge points, which occur at certain points
along the pipeline depending on the metadata columns defined in the TSV file.
As Nextflow will automatically download files from URLs, profiles and/or TSV
files, users can include links to publicly available data (e.g. the European
Bioinformatics Institutes’s ENA FTP server). This assists in reproducibility,
because if profiles or TSV files are uploaded with a publication, a researcher
wishing to re-analyse the data in the same way can use the exact settings and
file merging procedures in the original publication, without having to
reconstruct this from prose.
Monitoring
Users can either monitor their pipeline execution with the messages Nextflow
prints to the console while running, or utilise companion tools such as
Nextflow’s Tower [50] to monitor their analysis pipeline
during runtime.
Output
The pipeline produces a multitude of output files in various file formats, with
a more detailed listing available in the user documentation. These include
metrics, statistical analysis data, and standardised output files (BAM, VCF) for
close inspection and further downstream analysis, as well as a MultiQC report.
If an emailing daemon is set up on the server, the latter can be emailed to
users automatically, when starting the pipeline with a dedicated option
(--email you@yourdomain.org).
Benchmarking
Dual Screening of Human and Microbial Pathogen DNA
Full step-by-step instructions on the set up of the human and pathogen screening
demonstration (including input TSV file) can be seen in the supplementary
information. To demonstrate the efficiency and conciseness of nf-core/eager
pipeline in it’s dual role for both human and microbial screening of ancient
material, we replicated the results of Barquera et al. 2020
[81] using v2.2.0 (commit: e7471a7 and
Nextflow version: 20.04.1).
The following command was used to run the pipeline on the in-house servers at
the MPI-SHH, including a 2 TB memory node for running MALT against the NCBI Nt
(Nucleotide) database, and therefore the centralised custom profile for this
cluster was used.
To include the HOPS results from metagenomic screening in the report, we also
re-ran MultiQC with the upcoming version v1.10 (to be integrated into
nf-core/eager on release). After then installing the development version of
MultiQC (commit: 7584e64), as described in the MultiQC documentation
[86], we ran the following command in the results
directory of the nf-core/eager run, using the same configuration file.
Until MultiQC v1.10 is released, the HOPS heatmap is exported by nf-core/eager
in the corresponding MaltExtract results directory. Reports from both versions
(and the standalone HOPS PDF) can be seen in the supplementary information.
Pipeline Comparison
Full step-by-step instructions on the set up of the pipeline run-time
benchmarking, including environment and tool versions, can be seen in the
supplementary information. EAGER (v1.92.37) and nf-core/eager (v2.2.0,
commit: 830c22d; Nextflow v20.04.1) used the provided pre-built singularity
containers for software environments, whereas for PALEOMIX (v1.2.14) we
generated a custom conda environment (see supplementary information for the
environmental.yaml file). Run time comparisons were performed on a 32 CPU (AMD
Opteron 23xx) and 256 GB memory Red Hat QEMU Virtual Machine running the Ubuntu
18.04 operating system (Linux Kernel 4.15.0-112). Resource parameters of each
tool were only modified to specify the maximum available on the server and
otherwise left as default.
The following commands were used for each pipeline, with the commands run 10
times, each after cleaning up reference and results directories using a for
loop. Run times of the run commands themselves were measured using GNU Time.
## EAGER - description of XML files can be seen in supplementary informationsingularity exec \-B ~/benchmarks/output/EAGER:/data ~/.singularity/cache/EAGER-cache/EAGER-GUI_latest.sif \eagercli \/data## PALEOMIX - description of input YAML files can be seen in supplementary## informationpaleomix bam_pipeline run ~/benchmarks/output/paleomix/makefile_paleomix.yaml## paleomix optimised - description of input YAML files can be seen in## supplementary informationpaleomix bam_pipeline \run ~/benchmarks/output/paleomix_optimised/makefile_paleomix.yaml \--bwa-max-threads 4## nf-core/eager - description of resources configuration file (-c) can be seen## in supplementary informationnextflow run nf-core/eager -r 2.2.0 \--input ~/benchmarks/output/nfcore-eager-optimised/nfcore-eager_tsv.tsv \-c ~/.nextflow/pub_eager_vikingfish.conf \-profile pub_eager_vikingfish_optimised,pub_eager_vikingfish,singularity \--fasta ~/benchmarks/reference/GCF_902167405.1_gadMor3.0_genomic.fasta \--outdir ~/benchmarks/output/nfcore-eager-optimised/results/ \-w ~/benchmarks/output/nfcore-eager-optimised/work/ \--skip_fastqc \--skip_preseq \--run_bam_filtering \--bam_mapping_quality_threshold 25 \--bam_discard_unmapped \--bam_unmapped_type 'discard' \--dedupper 'markduplicates'##nf-core/eager optimised - description of resources profile(s) with optimised## bwa threads setting can be seen in supplementary informationnextflow run nf-core/eager -r 2.2.0 \--input ~/benchmarks/output/nfcore-eager-optimised/nfcore-eager_tsv.tsv \-c ~/.nextflow/pub_eager_vikingfish.conf \-profile pub_eager_vikingfish_optimised,pub_eager_vikingfish,singularity \--fasta ~/benchmarks/reference/GCF_902167405.1_gadMor3.0_genomic.fasta \--outdir ~/benchmarks/output/nfcore-eager-optimised/results/ \-w ~/benchmarks/output/nfcore-eager-optimised/work/ \--skip_fastqc \--skip_preseq \--run_bam_filtering \--bam_mapping_quality_threshold 25 \--bam_discard_unmapped \--bam_unmapped_type 'discard' \--dedupper 'markduplicates'
Mapping results across all pipelines showed very similar values, with low
variation across replicates as can be seen in Table 3.
Table 3: Comparison of common results values of key high-throughput short-read
data processing and mapping steps across the three pipelines. ‘qf’ stands for
mapping-quality filtered reads. All values represent mean and standard deviation
across 10 replicates of each pipeline, calculated from the output of the GNU
time tool.
sample_name
category
EAGER
nf-core/eager
PALEOMIX
COD076
processed_reads
71388991 ± 0
71388991 ± 0
72100142 ± 0
COD092
processed_reads
69615709 ± 0
69615709 ± 0
70249181 ± 0
COD076
mapped_qf_reads
16786467.7 ± 106.5
16786491.1 ± 89.9
16686607.2 ± 91.3
COD092
mapped_qf_reads
16283216.3 ± 71.3
16283194.7 ± 37.4
16207986.2 ± 44.4
COD076
ontarget_qf
23.5 ± 0
23.5 ± 0
23.1 ± 0
COD092
ontarget_qf
23.4 ± 0
23.4 ± 0
23.1 ± 0
COD076
dedupped_mapped_reads
12107264.4 ± 87.8
12107293.7 ± 69.7
12193415.8 ± 86.7
COD092
dedupped_mapped_reads
13669323.7 ± 87.6
13669328 ± 32.4
13795703.3 ± 47.9
COD076
mean_depth_coverage
0.9 ± 0
0.9 ± 0
0.9 ± 0
COD092
mean_depth_coverage
1 ± 0
1 ± 0
1 ± 0
COD076
mean_read_length
49.4 ± 0
49.4 ± 0
49.4 ± 0
COD092
mean_read_length
48.8 ± 0
48.8 ± 0
48.7 ± 0
Data and software availability
All pipeline code is available on GitHub at
https://github.com/nf-core/eager and
archived with Zenodo under the DOI
10.5281/zenodo.1465061. The version of
nf-core/eager that this manuscript is based on was the ‘dev’ branch of the GitHub
repository (2.2.0dev), and was released as v2.2.0. Demonstration data for dual
ancient human and pathogen screening from Barquera et al. [81] is
publicly available on the European Nucleotide Archive (ENA) under project
accession PRJEB37490. The human reference genome (hs37d5) and screening database
(Nucleotide or ‘nt’, October 2017) was downloaded from National Center for
Biotechnology Information FTP server. Ancient Cod genomic data from Star et al.
[4] used for benchmarking is publicly available on
the ENA under project accession PRJEB20524. The Gadus morhua reference genome
NCBI accession ID is: GCF_902167405.1.
This paper was collaboratively written with Manubot
[87], and supplementary information including
demonstration and benchmarking environments descriptions and walk-through can be
seen on GitHub at
https://github.com/apeltzer/eager2-paper/
and the supplement/ directory.
Competing Interests
No competing interests are declared.
Acknowledgements
We thank the nf-core community for general support and suggestions during the
writing of the pipeline. We also thank Arielle Munters, Hester van Schalkwyk,
Irina Velsko, Katherine Eaton, Luc Venturini, Marcel Keller, Pierre Lindenbaum,
Pontus Skoglund, Raphael Eisenhofer, Torsten Günter, Kevin Lord, and Åshild
Vågene for bug reports and feature suggestions. We are grateful to the members
of the Department of Archaeogenetics at the Max Planck Institute for the Science
of Human History who performed beta testing of the pipeline. We thank the aDNA
twitter community for responding to polls regarding design decisions during
development.
The Gesellschaft für wissenschaftliche Datenverarbeitung (GWDG, Göttingen)
kindly provided computational infrastructure for benchmarking. We also want to
thank Selina Carlhoff, Maria Spyrou, Elisabeth Nelson, Alexander Herbig and
Wolfgang Haak for providing comments and suggestions on this manuscript, and
acknowledge Christina Warinner, Stephan Schiffels and the Max Planck Society who
provided funds for travel to nf-core events. This project was also supported by
the ERC Starting Grant project (FoodTransforms) ERC-2015-StG 678901 funded by
the European Research Council awarded to Philipp W. Stockhammer (Ludwig
Maximilian University, Munich).
References
1. Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth
Eleftheria Palkopoulou, Swapan Mallick, Pontus Skoglund, Jacob Enk, Nadin Rohland, Heng Li, Ayça Omrak, Sergey Vartanyan, Hendrik Poinar, Anders Götherström, … Love Dalén Current Biology (2015-05) https://doi.org/34d
DOI: 10.1016/j.cub.2015.04.007 · PMID: 25913407 · PMCID: PMC4439331
2. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse
Ludovic Orlando, Aurélien Ginolhac, Guojie Zhang, Duane Froese, Anders Albrechtsen, Mathias Stiller, Mikkel Schubert, Enrico Cappellini, Bent Petersen, Ida Moltke, … Eske Willerslev Nature (2013-06-26) https://doi.org/q7n
DOI: 10.1038/nature12323 · PMID: 23803765
3. Ancient pigs reveal a near-complete genomic turnover following their introduction to Europe
Laurent A. F. Frantz, James Haile, Audrey T. Lin, Amelie Scheu, Christina Geörg, Norbert Benecke, Michelle Alexander, Anna Linderholm, Victoria E. Mullin, Kevin G. Daly, … Greger Larson Proceedings of the National Academy of Sciences (2019-08-27) https://doi.org/gf9hnf
DOI: 10.1073/pnas.1901169116 · PMID: 31405970 · PMCID: PMC6717267
4. Ancient DNA reveals the Arctic origin of Viking Age cod from Haithabu, Germany
Bastiaan Star, Sanne Boessenkool, Agata T. Gondek, Elena A. Nikulina, Anne Karin Hufthammer, Christophe Pampoulie, Halvor Knutsen, Carl André, Heidi M. Nistelberger, Jan Dierking, … James H. Barrett Proceedings of the National Academy of Sciences (2017-08-22) https://doi.org/gbt8b2
DOI: 10.1073/pnas.1710186114 · PMID: 28784790 · PMCID: PMC5576834
5. 137 ancient human genomes from across the Eurasian steppes
Peter de Barros Damgaard, Nina Marchi, Simon Rasmussen, Michaël Peyrot, Gabriel Renaud, Thorfinn Korneliussen, J. Víctor Moreno-Mayar, Mikkel Winther Pedersen, Amy Goldberg, Emma Usmanova, … Eske Willerslev Nature (2018-05-09) https://doi.org/gd8hs5
DOI: 10.1038/s41586-018-0094-2 · PMID: 29743675
6. A Draft Sequence of the Neandertal Genome
R. E. Green, J. Krause, A. W. Briggs, T. Maricic, U. Stenzel, M. Kircher, N. Patterson, H. Li, W. Zhai, M. H. Y. Fritz, … S. Paabo Science (2010-05-06) https://doi.org/c2x
DOI: 10.1126/science.1188021 · PMID: 20448178 · PMCID: PMC5100745
7. A High-Coverage Genome Sequence from an Archaic Denisovan Individual
M. Meyer, M. Kircher, M.-T. Gansauge, H. Li, F. Racimo, S. Mallick, J. G. Schraiber, F. Jay, K. Prufer, C. de Filippo, … S. Paabo Science (2012-08-30) https://doi.org/q8p
DOI: 10.1126/science.1224344 · PMID: 22936568 · PMCID: PMC3617501
8. The genome of the offspring of a Neanderthal mother and a Denisovan father
Viviane Slon, Fabrizio Mafessoni, Benjamin Vernot, Cesare de Filippo, Steffi Grote, Bence Viola, Mateja Hajdinjak, Stéphane Peyrégne, Sarah Nagel, Samantha Brown, … Svante Pääbo Nature (2018-08-22) https://doi.org/cs64
DOI: 10.1038/s41586-018-0455-x · PMID: 30135579 · PMCID: PMC6130845
9. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis
Kirsten I. Bos, Kelly M. Harkins, Alexander Herbig, Mireia Coscolla, Nico Weber, Iñaki Comas, Stephen A. Forrest, Josephine M. Bryant, Simon R. Harris, Verena J. Schuenemann, … Johannes Krause Nature (2014-08-20) https://doi.org/f6nk4g
DOI: 10.1038/nature13591 · PMID: 25141181 · PMCID: PMC4550673
10. Integrative approach using Yersinia pestis genomes to revisit the historical landscape of plague during the Medieval Period
Amine Namouchi, Meriam Guellil, Oliver Kersten, Stephanie Hänsch, Claudio Ottoni, Boris V. Schmid, Elsa Pacciani, Luisa Quaglia, Marco Vermunt, Egil L. Bauer, … Barbara Bramanti Proceedings of the National Academy of Sciences (2018-12-11) https://doi.org/ggfn3h
DOI: 10.1073/pnas.1812865115 · PMID: 30478041 · PMCID: PMC6294933
11. Ancient genomes reveal a high diversity of Mycobacterium leprae in medieval Europe
Verena J. Schuenemann, Charlotte Avanzi, Ben Krause-Kyora, Alexander Seitz, Alexander Herbig, Sarah Inskip, Marion Bonazzi, Ella Reiter, Christian Urban, Dorthe Dangvard Pedersen, … Johannes Krause PLOS Pathogens (2018-05-10) https://doi.org/gdrj4v
DOI: 10.1371/journal.ppat.1006997 · PMID: 29746563 · PMCID: PMC5944922
12. Ancient hepatitis B viruses from the Bronze Age to the Medieval period
Barbara Mühlemann, Terry C. Jones, Peter de Barros Damgaard, Morten E. Allentoft, Irina Shevnina, Andrey Logvin, Emma Usmanova, Irina P. Panyushkina, Bazartseren Boldgiv, Tsevel Bazartseren, … Eske Willerslev Nature (2018-05-09) https://doi.org/gddxvj
DOI: 10.1038/s41586-018-0097-z · PMID: 29743673
13. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B
Ben Krause-Kyora, Julian Susat, Felix M Key, Denise Kühnert, Esther Bosse, Alexander Immel, Christoph Rinne, Sabin-Christin Kornell, Diego Yepes, Sören Franzenburg, … Johannes Krause eLife (2018-05-10) https://doi.org/gdhck2
DOI: 10.7554/elife.36666 · PMID: 29745896 · PMCID: PMC6008052
14. Ancient reveals the timing and persistence of organellar genetic bottlenecks over 3,000 years of sunflower domestication and improvement
Nathan Wales, Melis Akman, Ray H. B. Watson, Fátima Sánchez Barreiro, Bruce D. Smith, Kristen J. Gremillion, M. Thomas P. Gilbert, Benjamin K. Blackman Evolutionary Applications (2018-02-13) https://doi.org/gf568v
DOI: 10.1111/eva.12594 · PMID: 30622634 · PMCID: PMC6304678
15. The origins and adaptation of European potatoes reconstructed from historical genomes
Rafal M. Gutaker, Clemens L. Weiß, David Ellis, Noelle L. Anglin, Sandra Knapp, José Luis Fernández-Alonso, Salomé Prat, Hernán A. Burbano Nature Ecology & Evolution (2019-06-24) https://doi.org/ggxkk8
DOI: 10.1038/s41559-019-0921-3 · PMID: 31235927
16. The Prevotella copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations
Adrian Tett, Kun D. Huang, Francesco Asnicar, Hannah Fehlner-Peach, Edoardo Pasolli, Nicolai Karcher, Federica Armanini, Paolo Manghi, Kevin Bonham, Moreno Zolfo, … Nicola Segata Cell Host & Microbe (2019-11) https://doi.org/ggc9dc
DOI: 10.1016/j.chom.2019.08.018 · PMID: 31607556 · PMCID: PMC6854460
17. CoproID predicts the source of coprolites and paleofeces using microbiome composition and host DNA content
Maxime Borry, Bryan Cordova, Angela Perri, Marsha Wibowo, Tanvi Prasad Honap, Jada Ko, Jie Yu, Kate Britton, Linus Girdland-Flink, Robert C. Power, … Christina Warinner PeerJ (2020-04-17) https://doi.org/dr8x
DOI: 10.7717/peerj.9001 · PMID: 32337106 · PMCID: PMC7169968
18. Pathogens and host immunity in the ancient human oral cavity
Christina Warinner, João F Matias Rodrigues, Rounak Vyas, Christian Trachsel, Natallia Shved, Jonas Grossmann, Anita Radini, Y Hancock, Raul Y Tito, Sarah Fiddyment, … Enrico Cappellini Nature Genetics (2014-02-23) https://doi.org/r4n
DOI: 10.1038/ng.2906 · PMID: 24562188 · PMCID: PMC3969750
19. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus
Laura S. Weyrich, Sebastian Duchene, Julien Soubrier, Luis Arriola, Bastien Llamas, James Breen, Alan G. Morris, Kurt W. Alt, David Caramelli, Veit Dresely, … Alan Cooper Nature (2017-03-08) https://doi.org/f9szrm
DOI: 10.1038/nature21674 · PMID: 28273061
20. Fifty thousand years of Arctic vegetation and megafaunal diet
Eske Willerslev, John Davison, Mari Moora, Martin Zobel, Eric Coissac, Mary E. Edwards, Eline D. Lorenzen, Mette Vestergård, Galina Gussarova, James Haile, … Pierre Taberlet Nature (2014-02-05) https://doi.org/f2zr4s
DOI: 10.1038/nature12921 · PMID: 24499916
21. Neandertal and Denisovan DNA from Pleistocene sediments
Viviane Slon, Charlotte Hopfe, Clemens L. Weiß, Fabrizio Mafessoni, Marco de la Rasilla, Carles Lalueza-Fox, Antonio Rosas, Marie Soressi, Monika V. Knul, Rebecca Miller, … Matthias Meyer Science (2017-05-12) https://doi.org/b6jd
DOI: 10.1126/science.aam9695 · PMID: 28450384
22. Plasmodium vivax Malaria Viewed through the Lens of an Eradicated European Strain
Lucy van Dorp, Pere Gelabert, Adrien Rieux, Marc de Manuel, Toni de-Dios, Shyam Gopalakrishnan, Christian Carøe, Marcela Sandoval-Velasco, Rosa Fregel, Iñigo Olalde, … Carles Lalueza-Fox Molecular Biology and Evolution (2020-03) https://doi.org/ggqzq2
DOI: 10.1093/molbev/msz264 · PMID: 31697387 · PMCID: PMC7038659
23. Paging through history: parchment as a reservoir of ancient DNA for next generation sequencing
M. D. Teasdale, N. L. van Doorn, S. Fiddyment, C. C. Webb, T. O’Connor, M. Hofreiter, M. J. Collins, D. G. Bradley Philosophical Transactions of the Royal Society B: Biological Sciences (2015-01-19) https://doi.org/ggqzq3
DOI: 10.1098/rstb.2013.0379 · PMID: 25487331 · PMCID: PMC4275887
24. A 5700 year-old human genome and oral microbiome from chewed birch pitch
Theis Z. T. Jensen, Jonas Niemann, Katrine Højholt Iversen, Anna K. Fotakis, Shyam Gopalakrishnan, Åshild J. Vågene, Mikkel Winther Pedersen, Mikkel-Holger S. Sinding, Martin R. Ellegaard, Morten E. Allentoft, … Hannes Schroeder Nature Communications (2019-12-17) https://doi.org/ggfm6x
DOI: 10.1038/s41467-019-13549-9 · PMID: 31848342 · PMCID: PMC6917805
25. Ancient DNA from mastics solidifies connection between material culture and genetics of mesolithic hunter–gatherers in Scandinavia
Natalija Kashuba, Emrah Kırdök, Hege Damlien, Mikael A. Manninen, Bengt Nordqvist, Per Persson, Anders Götherström Communications Biology (2019-05-15) https://doi.org/ggqzqz
DOI: 10.1038/s42003-019-0399-1 · PMID: 31123709 · PMCID: PMC6520363
26. The Beaker phenomenon and the genomic transformation of northwest Europe
Iñigo Olalde, Selina Brace, Morten E. Allentoft, Ian Armit, Kristian Kristiansen, Thomas Booth, Nadin Rohland, Swapan Mallick, Anna Szécsényi-Nagy, Alissa Mittnik, … David Reich Nature (2018-02-21) https://doi.org/gcx74m
DOI: 10.1038/nature25738 · PMID: 29466337 · PMCID: PMC5973796
27. The genomic history of southeastern Europe
Iain Mathieson, Songül Alpaslan-Roodenberg, Cosimo Posth, Anna Szécsényi-Nagy, Nadin Rohland, Swapan Mallick, Iñigo Olalde, Nasreen Broomandkhoshbacht, Francesca Candilio, Olivia Cheronet, … David Reich Nature (2018-02-21) https://doi.org/gc2n9h
DOI: 10.1038/nature25778 · PMID: 29466330 · PMCID: PMC6091220
28. A draft genome of Yersinia pestis from victims of the Black Death
Kirsten I. Bos, Verena J. Schuenemann, G. Brian Golding, Hernán A. Burbano, Nicholas Waglechner, Brian K. Coombes, Joseph B. McPhee, Sharon N. DeWitte, Matthias Meyer, Sarah Schmedes, … Johannes Krause Nature (2011-10-12) https://doi.org/fk87wk
DOI: 10.1038/nature10549 · PMID: 21993626 · PMCID: PMC3690193
29. Bioinformatics Education–Perspectives and Challenges out of Africa
O. Tastan Bishop, E. F. Adebiyi, A. M. Alzohairy, D. Everett, K. Ghedira, A. Ghouila, J. Kumuthini, N. J. Mulder, S. Panji, H.-G. Patterton, (for the H3ABioNet Consortium, as members of The H3Africa Consortium) Briefings in Bioinformatics (2014-07-02) https://doi.org/f67hjx
DOI: 10.1093/bib/bbu022 · PMID: 24990350 · PMCID: PMC4364068
30. Highlights on the Application of Genomics and Bioinformatics in the Fight Against Infectious Diseases: Challenges and Opportunities in Africa
Saikou Y. Bah, Collins Misita Morang’a, Jonas A. Kengne-Ouafo, Lucas Amenga–Etego, Gordon A. Awandare Frontiers in Genetics (2018-11-27) https://doi.org/gfrxbz
DOI: 10.3389/fgene.2018.00575 · PMID: 30538723 · PMCID: PMC6277583
32. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins
Matthias Meyer, Juan-Luis Arsuaga, Cesare de Filippo, Sarah Nagel, Ayinuer Aximu-Petri, Birgit Nickel, Ignacio Martínez, Ana Gracia, José María Bermúdez de Castro, Eudald Carbonell, … Svante Pääbo Nature (2016-03-14) https://doi.org/bdcn
DOI: 10.1038/nature17405 · PMID: 26976447
33. Patterns of damage in genomic DNA sequences from a Neandertal
A. W. Briggs, U. Stenzel, P. L. F. Johnson, R. E. Green, J. Kelso, K. Prufer, M. Meyer, J. Krause, M. T. Ronan, M. Lachmann, S. Paabo Proceedings of the National Academy of Sciences (2007-08-21) https://doi.org/bs4w7h
DOI: 10.1073/pnas.0704665104 · PMID: 17715061 · PMCID: PMC1976210
34. A new model for ancient DNA decay based on paleogenomic meta-analysis
Logan Kistler, Roselyn Ware, Oliver Smith, Matthew Collins, Robin G. Allaby Nucleic Acids Research (2017-06-20) https://doi.org/gf58ts
DOI: 10.1093/nar/gkx361 · PMID: 28486705 · PMCID: PMC5499742
35. A Robust Framework for Microbial Archaeology
Christina Warinner, Alexander Herbig, Allison Mann, James A. Fellows Yates, Clemens L. Weiß, Hernán A. Burbano, Ludovic Orlando, Johannes Krause Annual Review of Genomics and Human Genetics (2017-08-31) https://doi.org/gf5wqv
DOI: 10.1146/annurev-genom-091416-035526 · PMID: 28460196 · PMCID: PMC5581243
36. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters
Hákon Jónsson, Aurélien Ginolhac, Mikkel Schubert, Philip L. F. Johnson, Ludovic Orlando Bioinformatics (2013-07) https://doi.org/gb5g2t
DOI: 10.1093/bioinformatics/btt193 · PMID: 23613487 · PMCID: PMC3694634
37. Early Divergent Strains of Yersinia pestis in Eurasia 5,000 Years Ago
Simon Rasmussen, Morten Erik Allentoft, Kasper Nielsen, Ludovic Orlando, Martin Sikora, Karl-Göran Sjögren, Anders Gorm Pedersen, Mikkel Schubert, Alex Van Dam, Christian Moliin Outzen Kapel, … Eske Willerslev Cell (2015-10) https://doi.org/f3mxqd
DOI: 10.1016/j.cell.2015.10.009 · PMID: 26496604 · PMCID: PMC4644222
38. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX
Mikkel Schubert, Luca Ermini, Clio Der Sarkissian, Hákon Jónsson, Aurélien Ginolhac, Robert Schaefer, Michael D Martin, Ruth Fernández, Martin Kircher, Molly McCue, … Ludovic Orlando Nature Protocols (2014-04-10) https://doi.org/f5x3qm
DOI: 10.1038/nprot.2014.063 · PMID: 24722405
39. EAGER: efficient ancient genome reconstruction
Alexander Peltzer, Günter Jäger, Alexander Herbig, Alexander Seitz, Christian Kniep, Johannes Krause, Kay Nieselt Genome Biology (2016-03-31) https://doi.org/ggqzpk
DOI: 10.1186/s13059-016-0918-z · PMID: 27036623 · PMCID: PMC4815194
41. mapDamage: testing for damage patterns in ancient DNA sequences
Aurelien Ginolhac, Morten Rasmussen, M. Thomas P. Gilbert, Eske Willerslev, Ludovic Orlando Bioinformatics (2011-08-01) https://doi.org/cn45v7
DOI: 10.1093/bioinformatics/btr347 · PMID: 21659319
42. The Stone Age Plague and Its Persistence in Eurasia
Aida Andrades Valtueña, Alissa Mittnik, Felix M. Key, Wolfgang Haak, Raili Allmäe, Andrej Belinskij, Mantas Daubaras, Michal Feldman, Rimantas Jankauskas, Ivor Janković, … Johannes Krause Current Biology (2017-12) https://doi.org/cgmv
DOI: 10.1016/j.cub.2017.10.025 · PMID: 29174893
44. Nextflow enables reproducible computational workflows
Paolo Di Tommaso, Maria Chatzou, Evan W Floden, Pablo Prieto Barja, Emilio Palumbo, Cedric Notredame Nature Biotechnology (2017-04-11) https://doi.org/gfj52z
DOI: 10.1038/nbt.3820 · PMID: 28398311
45. The nf-core framework for community-curated bioinformatics pipelines
Philip A. Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso, Sven Nahnsen Nature Biotechnology (2020-02-13) https://doi.org/ggk3qh
DOI: 10.1038/s41587-020-0439-x · PMID: 32055031
51. Improving ancient DNA read mapping against modern reference genomes
Mikkel Schubert, Aurelien Ginolhac, Stinus Lindgreen, John F Thompson, Khaled AS AL-Rasheid, Eske Willerslev, Anders Krogh, Ludovic Orlando BMC Genomics (2012) https://doi.org/gb3ff7
DOI: 10.1186/1471-2164-13-178 · PMID: 22574660 · PMCID: PMC3468387
58. The Sequence Alignment/Map format and SAMtools
H. Li, B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis, R. Durbin, 1000 Genome Project Data Processing Subgroup Bioinformatics (2009-06-08) https://doi.org/ff6426
DOI: 10.1093/bioinformatics/btp352 · PMID: 19505943 · PMCID: PMC2723002
59. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data
A. McKenna, M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis, A. Kernytsky, K. Garimella, D. Altshuler, S. Gabriel, M. Daly, M. A. DePristo Genome Research (2010-07-19) https://doi.org/bnzbn6
DOI: 10.1101/gr.107524.110 · PMID: 20644199 · PMCID: PMC2928508
63. Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe
Thiseas C. Lamnidis, Kerttu Majander, Choongwon Jeong, Elina Salmela, Anna Wessman, Vyacheslav Moiseyev, Valery Khartanovich, Oleg Balanovsky, Matthias Ongyerth, Antje Weihmann, … Stephan Schiffels Nature Communications (2018-11-27) https://doi.org/ggxkk6
DOI: 10.1038/s41467-018-07483-5 · PMID: 30479341 · PMCID: PMC6258758
64. DamageProfiler: Fast damage pattern calculation for ancient DNA
Judith Neukamm, Alexander Peltzer, Kay Nieselt Cold Spring Harbor Laboratory (2020-10-01) https://doi.org/ghd45j
DOI: 10.1101/2020.10.01.322206
65. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal
Pontus Skoglund, Bernd H. Northoff, Michael V. Shunkov, Anatoli P. Derevianko, Svante Pääbo, Johannes Krause, Mattias Jakobsson Proceedings of the National Academy of Sciences (2014-02-11) https://doi.org/f2z5sw
DOI: 10.1073/pnas.1318934111 · PMID: 24469802 · PMCID: PMC3926038
66. An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data
Goo Jun, Mary Kate Wing, Gonçalo R. Abecasis, Hyun Min Kang Genome Research (2015-06) https://doi.org/f7dz2d
DOI: 10.1101/gr.176552.114 · PMID: 25883319 · PMCID: PMC4448687
70. Bioconda: sustainable and comprehensive software distribution for the life sciences
Björn Grüning, Ryan Dale, Andreas Sjödin, Brad A. Chapman, Jillian Rowe, Christopher H. Tomkins-Tinch, Renan Valieris, Johannes Köster, The Bioconda Team Nature Methods (2018-07-02) https://doi.org/gd2xzp
DOI: 10.1038/s41592-018-0046-7 · PMID: 29967506
72. Schmutzi: estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA
Gabriel Renaud, Viviane Slon, Ana T. Duggan, Janet Kelso Genome Biology (2015-10-12) https://doi.org/f72mvg
DOI: 10.1186/s13059-015-0776-0 · PMID: 26458810 · PMCID: PMC4601135
73. Assessing DNA Sequence Alignment Methods for Characterizing Ancient Genomes and Methylomes
Marine Poullet, Ludovic Orlando Frontiers in Ecology and Evolution (2020-05-06) https://doi.org/ggzwqr
DOI: 10.3389/fevo.2020.00105
75. Haplotype-based variant detection from short-read sequencing
Erik Garrison, Gabor Marth arXiv (2012-07-24) https://arxiv.org/abs/1207.3907
76. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman
Alexander Herbig, Frank Maixner, Kirsten I. Bos, Albert Zink, Johannes Krause, Daniel H. Huson bioRxiv (2016-04-27) https://doi.org/ggxkk9
DOI: 10.1101/050559
77. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico
Åshild J. Vågene, Alexander Herbig, Michael G. Campana, Nelly M. Robles García, Christina Warinner, Susanna Sabin, Maria A. Spyrou, Aida Andrades Valtueña, Daniel Huson, Noreen Tuross, … Johannes Krause Nature Ecology & Evolution (2018-01-15) https://doi.org/ggxkk7
DOI: 10.1038/s41559-017-0446-6 · PMID: 29335577
79. HOPS: automated detection and authentication of pathogen DNA in archaeological remains
Ron Hübler, Felix M. Key, Christina Warinner, Kirsten I. Bos, Johannes Krause, Alexander Herbig Genome Biology (2019-12-16) https://doi.org/ggxkmb
DOI: 10.1186/s13059-019-1903-0 · PMID: 31842945 · PMCID: PMC6913047
80. Microbial differences between dental plaque and historic dental calculus are related to oral biofilm maturation stage
Irina M. Velsko, James A. Fellows Yates, Franziska Aron, Richard W. Hagan, Laurent A. F. Frantz, Louise Loe, Juan Bautista Rodriguez Martinez, Eros Chaves, Chris Gosden, Greger Larson, Christina Warinner Microbiome (2019-07-06) https://doi.org/ggxkmc
DOI: 10.1186/s40168-019-0717-3 · PMID: 31279340 · PMCID: PMC6612086
81. Origin and Health Status of First-Generation Africans from Early Colonial Mexico
Rodrigo Barquera, Thiseas C. Lamnidis, Aditya Kumar Lankapalli, Arthur Kocher, Diana I. Hernández-Zaragoza, Elizabeth A. Nelson, Adriana C. Zamora-Herrera, Patxi Ramallo, Natalia Bernal-Felipe, Alexander Immel, … Johannes Krause Current Biology (2020-06) https://doi.org/ggwq88
DOI: 10.1016/j.cub.2020.04.002 · PMID: 32359431
83. ATLAS: Analysis Tools for Low-depth and Ancient Samples
Vivian Link, Athanasios Kousathanas, Krishna Veeramah, Christian Sell, Amelie Scheu, Daniel Wegmann Cold Spring Harbor Laboratory (2017-03-24) https://doi.org/gg668z
DOI: 10.1101/105346
87. Open collaborative writing with Manubot
Daniel S. Himmelstein, Vincent Rubinetti, David R. Slochower, Dongbo Hu, Venkat S. Malladi, Casey S. Greene, Anthony Gitter PLOS Computational Biology (2019-06-24) https://doi.org/c7np
DOI: 10.1371/journal.pcbi.1007128 · PMID: 31233491 · PMCID: PMC6611653
 0000-0001-5585-6277
·
0000-0001-5585-6277
·  jfy133
·
jfy133
·  jafellowsyates
jafellowsyates